|
Konghao Zhao
konghaoz at usc dot edu
Hello, my name is Konghao Zhao. I am a second-year PhD student in Computer Science at the University of Southern California, advised by Ruishan Liu in the Laboratory for Machine Learning, Health and Biomedicine.
My current research focuses on AI in healthcare, specifically on clinical trials designs and multi-modal analysis. I aim to design and implement high-performance, interpretable, and reliable ML/AI solutions for high-stakes human health applications.
I obtained a B.S. degree in CS and a B.A. degree in Math with Magna Cum Laude at Wake Forest University, where I was fortunate to conduct research as an undergraduate researcher at the Wake Forest DataMine Research Group advised by Natalia Khuri.
My research were primarily based on leveraging multi-objective optimization and ML/AI approaches to build solutions for applications related to single-cell RNA Sequencing data.
I interned at Boehringer Ingelheim in the Global Experimental Medicine Data Analysis and Biostatistics group in Fall 2025 and returned in Summer 2026, advised by Dacheng Liu, focusing on clinical data science for Phase I oncology clinical trial design.
Previously, I was a summer scholar at Carnegie Mellon Robotics Institute's Advanced Agent-Robotics Technology Lab, advised by Katia Sycara, where I worked on improving the performance and interpretability of Concept Bottleneck Models.
[ Email /
CV /
LinkedIn /
Google Scholar /
Github ]
|

|
Selected Publications
|
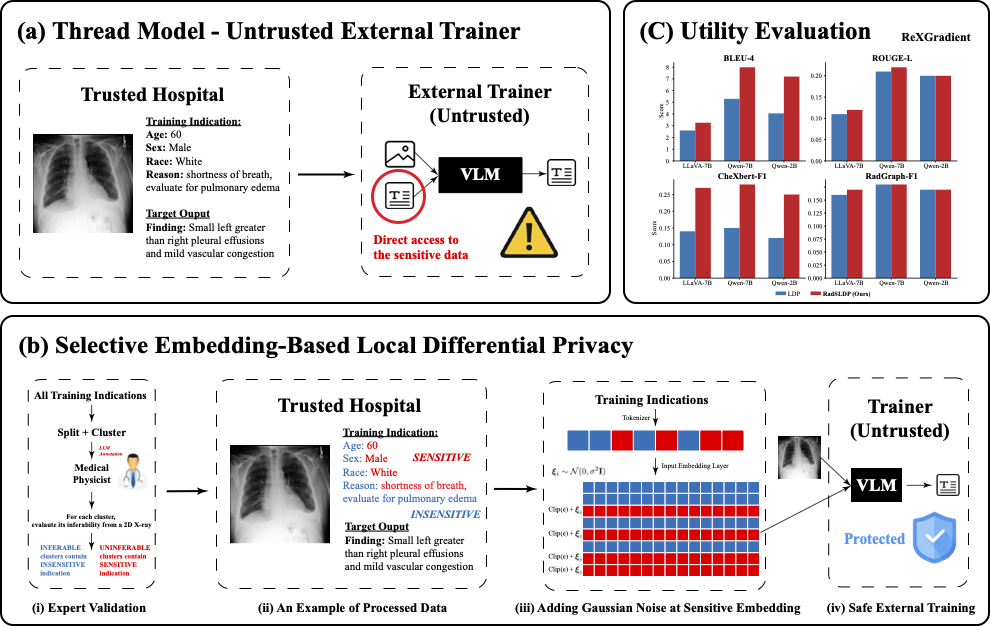
RadSLDP: Selective Local Differential Privacy for Radiology Vision-Language Models
Konghao Zhao, Xinyang Xu, Runhui Xu, Yutaka Natsuaki, Wenjie Xiong, Sai Praneeth Karimireddy, and Ruishan Liu
September 2026, Medical Image Computing and Computer-Assisted Intervention (MICCAI)
Regular Paper
▶ Show Abstract
Medical Vision–Language Models (VLMs) for radiology report generation are often fine-tuned by external developers due to hospitals’ limited computational infrastructure. It requires sensitive patient data to be transmitted to untrusted training environments, which creates privacy risks. Standard Differential Privacy protects the trained model but assumes trusted access to raw data, making it unsuitable for untrusted external training. Local Differential Privacy (LDP) enables privacy-preserving training in untrusted settings by perturbing all text token embeddings before transmission but significantly degrading clinical utility. We observe that clinical reports contain both image-inferable and non-inferable content and define the non-inferable portion as nonvisual clinical context (NVCC), such as demographics and medical history, which constitutes the primary privacy risk. To preserve privacy while maintaining utility, we propose RadSLDP, a selective LDP mechanism that perturbs only expert-validated NVCC token embeddings, with each protected token embedding satisfying formal per-token (ε, δ)-LDP via the Gaussian mechanism. As a result, RadSLDP matches or outperforms uniform LDP in 94% of comparisons, including 29/36 improvements and 5/36 ties. It improves BLEU-4 by 24–77% and CheXbert Micro-F1 by 87–108% on ReXGradient dataset. RadSLDP also exhibits lower performance variability across privacy budgets, enabling practical privacy-preserving VLM fine-tuning in untrusted settings.
|
|
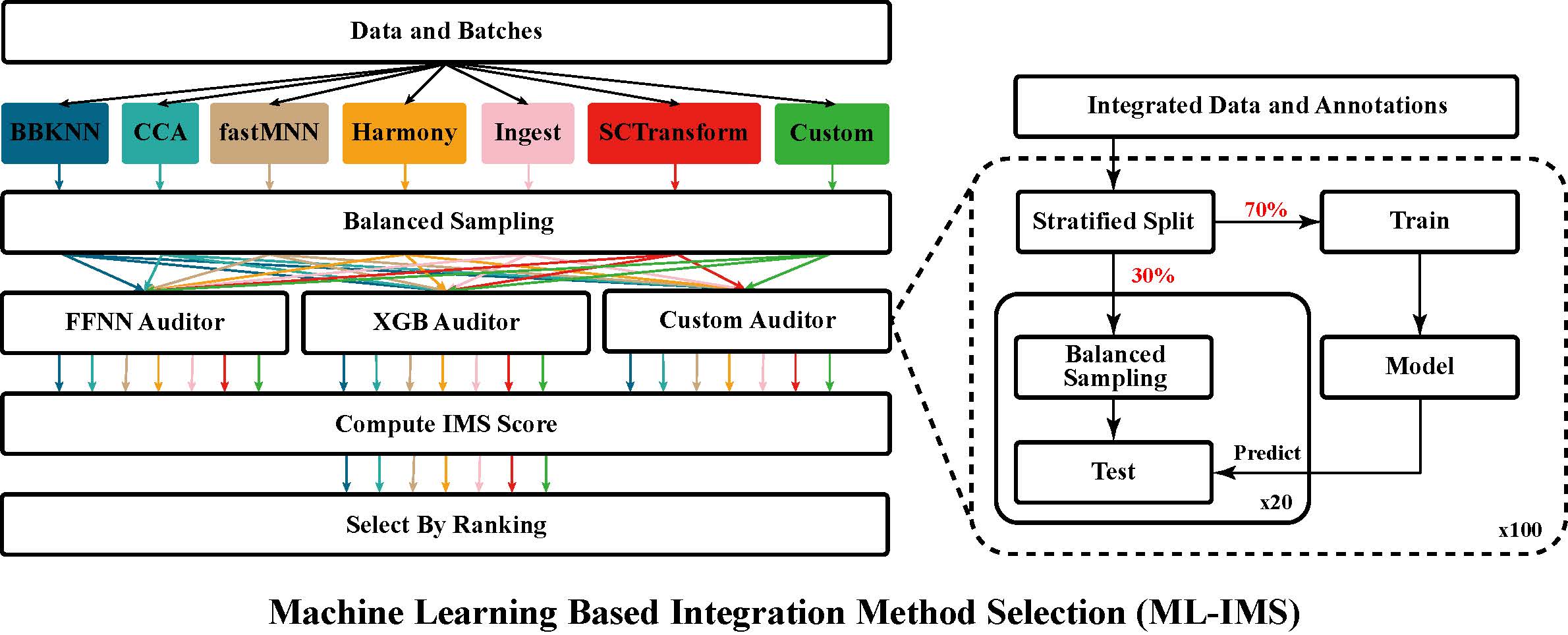
An Ensemble Machine Learning Approach for Benchmarking and Selection of scRNA-seq Integration Methods
Konghao Zhao, Sapan Bhandari, Nathan P. Whitener, Jason M. Grayson, and Natalia Khuri
September 2023, ACM-BCB
Top 10% Oral Presentation
▶ Show Abstract
Accurate integration of high-dimensional single-cell sequencing datasets is important for the construction of cell atlases and for the discovery of biomarkers. Because the performance of integration methods varies in different scenarios and on different datasets, it is important to provide end users with an automated system for the benchmarking and selection of the best integration among several alternatives. Here, we present a system that uses an ensemble of auditors, trained by supervised machine learning, which quantifies residual variability of integrated data and automatically selects the integration with the smallest difference between observed and expected batch effects. A rigorous and systematic validation was performed using 6 popular integration methods and 52 benchmark datasets. Algorithmic and data biases were uncovered and shortcomings of existing validation metrics were examined. Our results demonstrate the utility, validity, flexibility and consistency of the proposed approach.
|
|
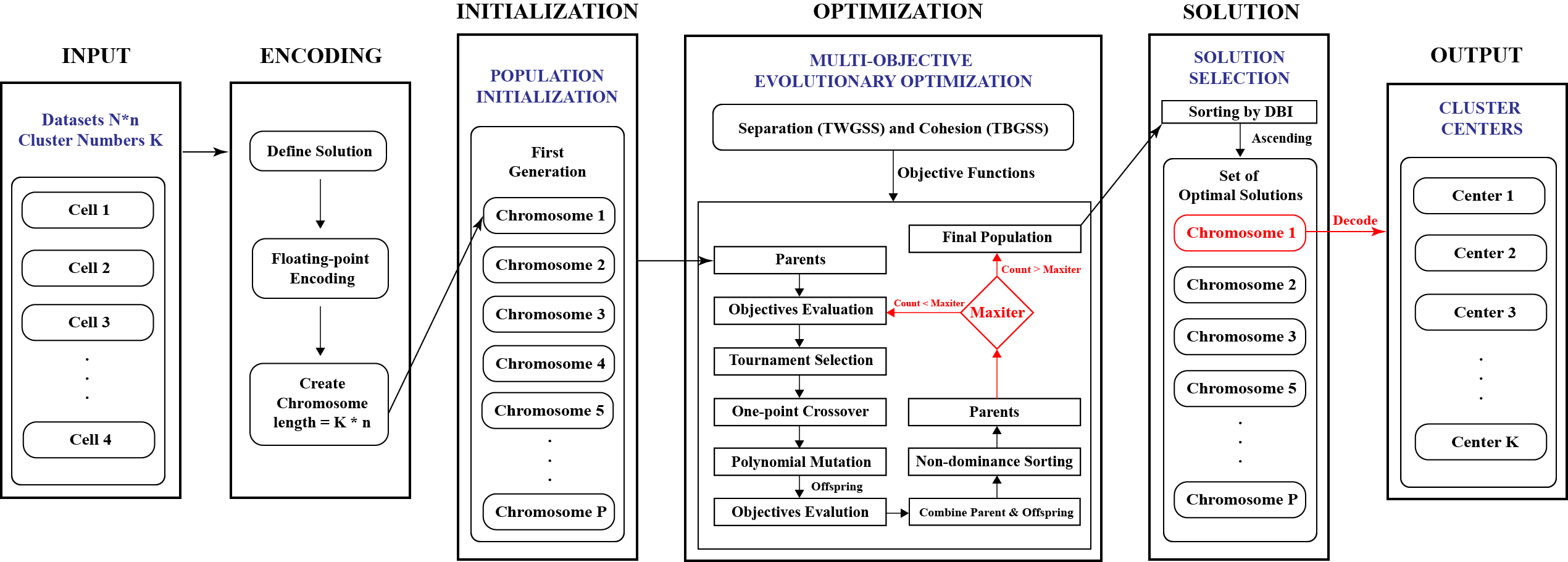
Multi-Objective Genetic Algorithm for Cluster Analysis of Single-Cell Transcriptomes
Konghao Zhao, Jason M. Grayson, and Natalia Khuri
January 2023, Journal of Personalized Medicine
Monthly Cover
▶ Show Abstract
Cells are the basic building blocks of human organisms, and the identification of their types and states in transcriptomic data is an important and challenging task. Many of the existing approaches to cell-type prediction are based on clustering methods that optimize only one criterion. In this paper, a multi-objective Genetic Algorithm for cluster analysis is proposed, implemented, and systematically validated on 48 experimental and 60 synthetic datasets. The results demonstrate that the performance and the accuracy of the proposed algorithm are reproducible, stable, and better than those of single-objective clustering methods. Computational run times of multi-objective clustering of large datasets were studied and used in supervised machine learning to accurately predict the execution times of clustering of new single-cell transcriptomes.
|
Additional Publications
|
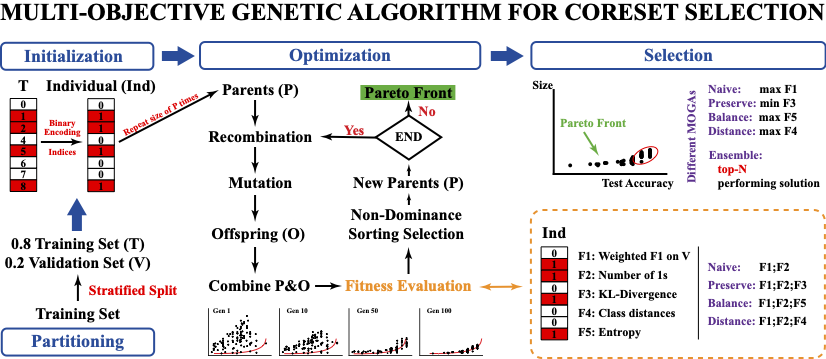
Multi-Objective Evolutionary Optimization for the Discovery of Structurally Diverse and Active Compounds
Konghao Zhao, Yimin Wang, O. Hernandez Segura, and Natalia Khuri
May 2026, ACM-BCB
Short Paper
▶ Show Abstract
Quantitative Structure-Activity Relationship (QSAR) is a computational method used in drug discovery that models relationships between compounds’ physico-chemical properties and activities. Historically, QSAR models were linear, which limited their use to structurally similar molecules and optimization of their properties. Today, complex and non-linear relationships can be modeled using machine learning (ML), and ML-QSAR models can efficiently classify compounds of massive and structurally diverse chemical libraries. Nevertheless, the applicability domain and accuracy of ML-QSAR models remains limited due to two major challenges. First, the data used for training of these models is imbalanced, with a majority of training compounds being inactive. A related issue is the small size of the training data set itself. Although the chemical space of synthesizable compounds is unlimited, experimental screening data, which is used for training of ML-QSAR, is often small in size. Combined with the imbalance, this makes it difficult to train robust and generalizable models that can accurately predict the activity of compounds that are structurally dissimilar from the training set. The second challenge is due to the intended use of ML-QSAR models. The goal of virtual screening is to identify a small, relevant subset of compounds for experimental validation, but the criteria for this selection can vary greatly. For example, some screening campaigns seek compounds that are structurally similar to approved drugs, while others focus on the discovery of novel structures. This paper addresses these issues by formulating the problem of data selection as a multi-objective optimization task, and solving it using a multi-objective Genetic Algorithm. This approach provides a flexible and systematic way to select subsets of data tailored for different needs, taking into consideration competing objectives like structural diversity and class balance. Our work demonstrates that this method outperforms existing multi-objective optimization approaches and identifies structurally diverse compounds in a largescale virtual screening of over 1 million chemicals.
|
|
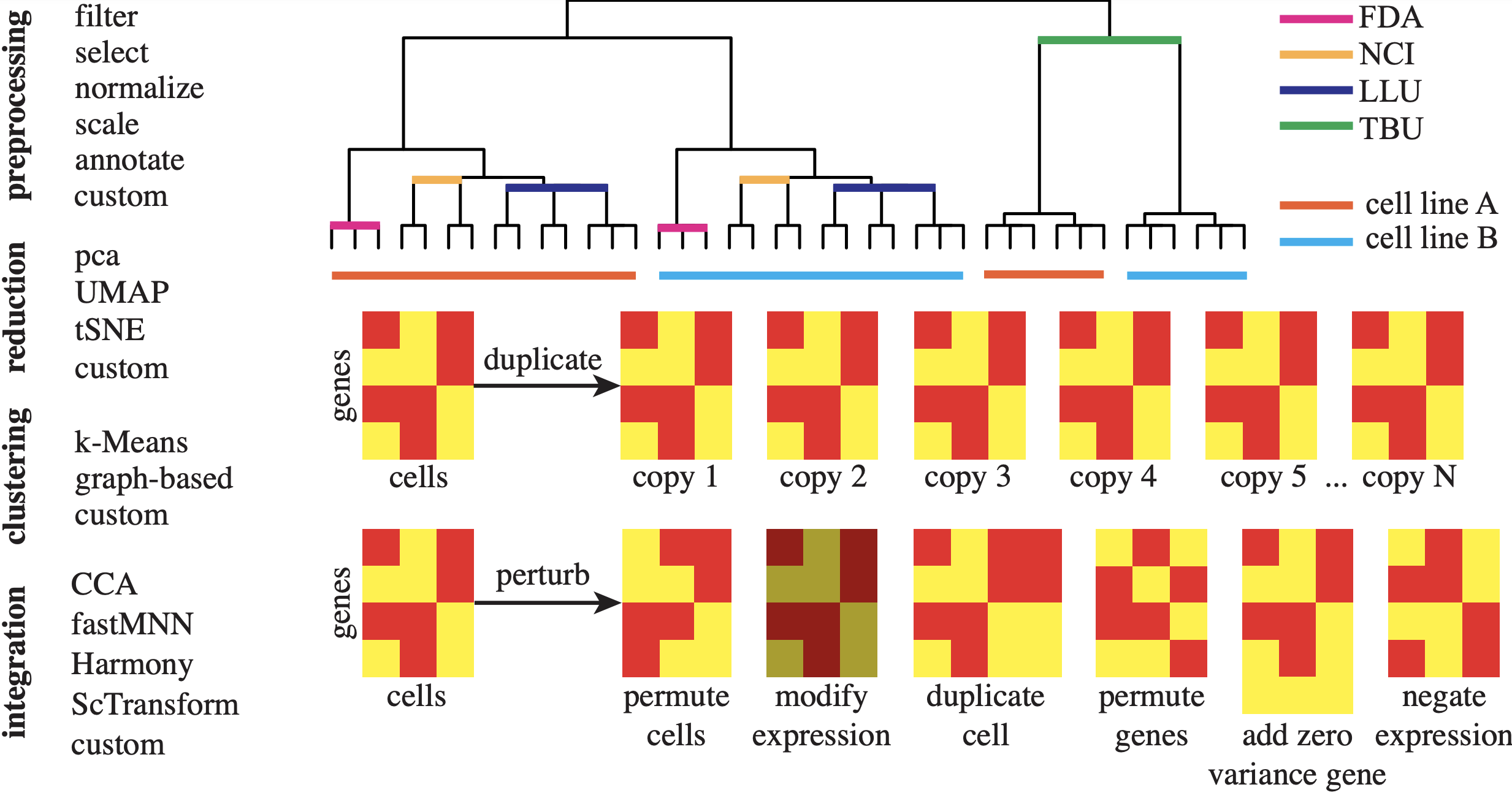
Data-Centric Framework for Testing and Benchmarking of scRNA-seq Pipelines
Konghao Zhao, Nathan P. Whitener, and Natalia Khuri
October 2025, AIHMA
Full Article
▶ Show Abstract
Computational methods for the analysis of single-cell RNA sequencing data are driving advances in personalized medicine. Given the rapid integration of these complex tools into research and development pipelines, the systematic and reliable testing of computational methods is critical to ensure the integrity of derived insights. However, a significant gap exists in appropriate benchmarks and innovative frameworks for the systematic and rigorous evaluation of existing and emergent digital technologies. In this work, we present a new framework, powered by a software package called scrnabench, to conduct systematic testing and benchmarking of computational tools. This framework supports the development and evaluation of digital health technologies by ensuring reliable, stable, and trustworthy results. The package can be used to select the most appropriate method for data analyses, to evaluate emergent tools, and to identify the strengths and weaknesses of existing software. In addition, we leverage software engineering techniques of metamorphic testing to help uncover implementation errors, faults, and anomalies and increase trust in AI-driven techniques. The scrnabench package is open-source, accessible and extendable, and we demonstrate its unique features in several use-case scenarios
|
|
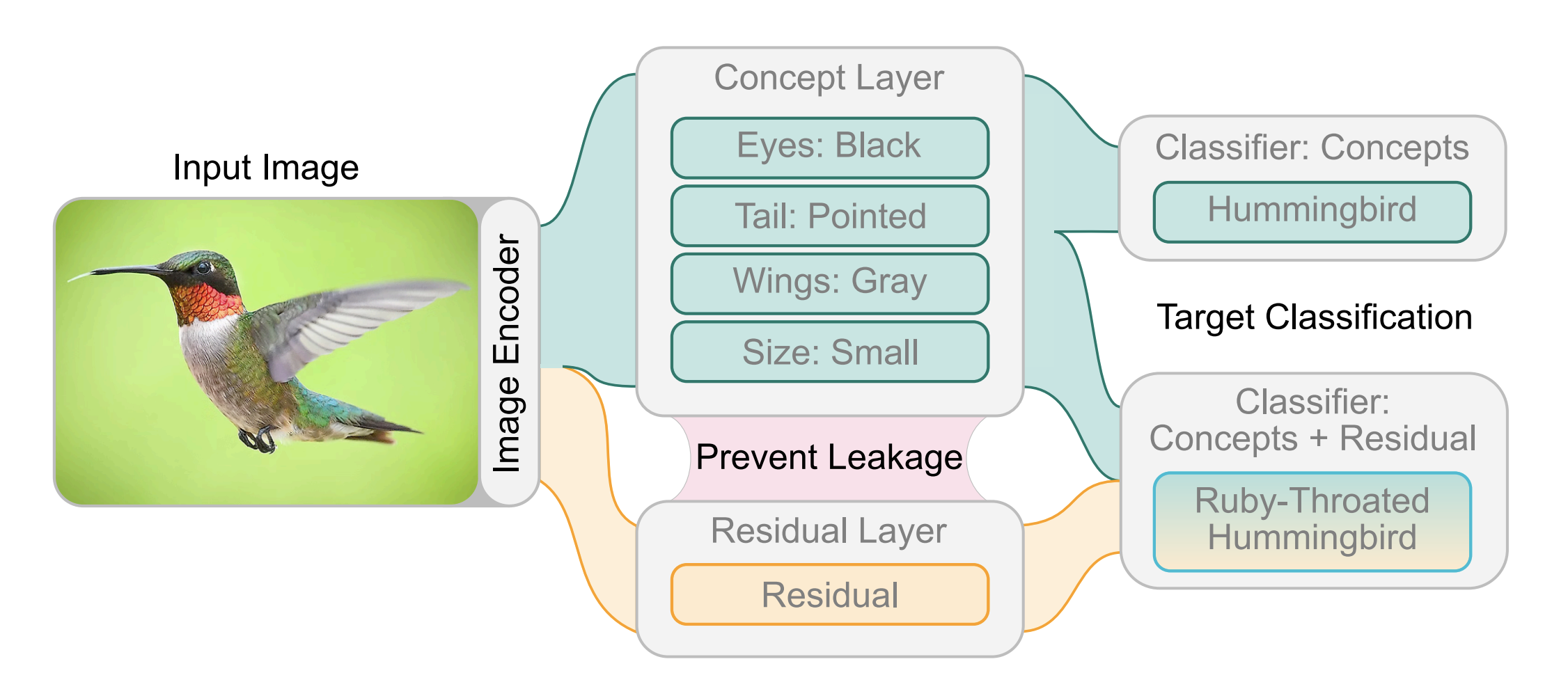
Disentangled Concept-Residual Models: Bridging the Interpretability–Performance Gap for Incomplete Concept Sets
Renos Zabounidis, Ini Oguntola, Konghao Zhao, Joseph Campbell, Simon Stepputtis, and Katia Sycara
December 2025, TMLR
Regular Paper
▶ Show Abstract
Deploying AI in high-stakes settings requires models that are not only accurate but also interpretable and amenable to human oversight. Concept Bottleneck Models (CBMs) support these goals by structuring predictions around human-understandable concepts, enabling interpretability and post-hoc human intervenability. However, CBMs rely on a ‘complete’ concept set, requiring practitioners to define and label enough concepts to match the predictive power of black-box models. To relax this requirement, prior work introduced residual connections that bypass the concept layer and recover information missing from an incomplete concept set. While effective in bridging the performance gap, these residuals can redundantly encode concept information, a phenomenon we term concept-residual overlap. In this work, we investigate the effects of concept-residual overlap and evaluate strategies to mitigate it. We (1) define metrics to quantify the extent of concept-residual overlap in CRMs; (2) introduce complementary metrics to evaluate how this overlap impacts interpretability, concept importance, and the effectiveness of concept-based interventions; and (3) present Disentangled Concept-Residual Models (D-CRMs), a general class of CRMs designed to mitigate this issue. Within this class, we propose a novel disentanglement approach based on minimizing mutual information (MI). Using CelebA, CIFAR100, AA2, CUB, and OAI, we show that standard CRMs exhibit significant concept-residual overlap, and that reducing this overlap with MI-based D-CRMs restores key properties of CBMs, including interpretability, functional reliance on concepts, and intervention robustness, without sacrificing predictive performance.
|

|
An Evolutionary Approach to Data Valuation
Sapan Bhandari, Nathan P. Whitener, Konghao Zhao, and Natalia Khuri
August 2022, ACM-BCB
Regular Paper
▶ Show Abstract
Data valuation in machine learning comprises computational methods for the estimation of the importance of individual training instances. It has been used to remove noise, uncover biases, and improve the accuracy of trained models. Current data valuation techniques do not scale up for large datasets and do not work for regression tasks, where the objective is to predict a numerical outcome rather than a small number of nominal class labels. In this work, an evolutionary approach for qualitative and quantitative data valuation, is presented. The proposed approach is tested on regression and classification benchmarks, and on several bioinformatics and health informatics datasets. In addition, models trained with most valuable subsets of data are validated on independently acquired tests, demonstrating the generalizability as well as the practical utility of the proposed approach.
|
|
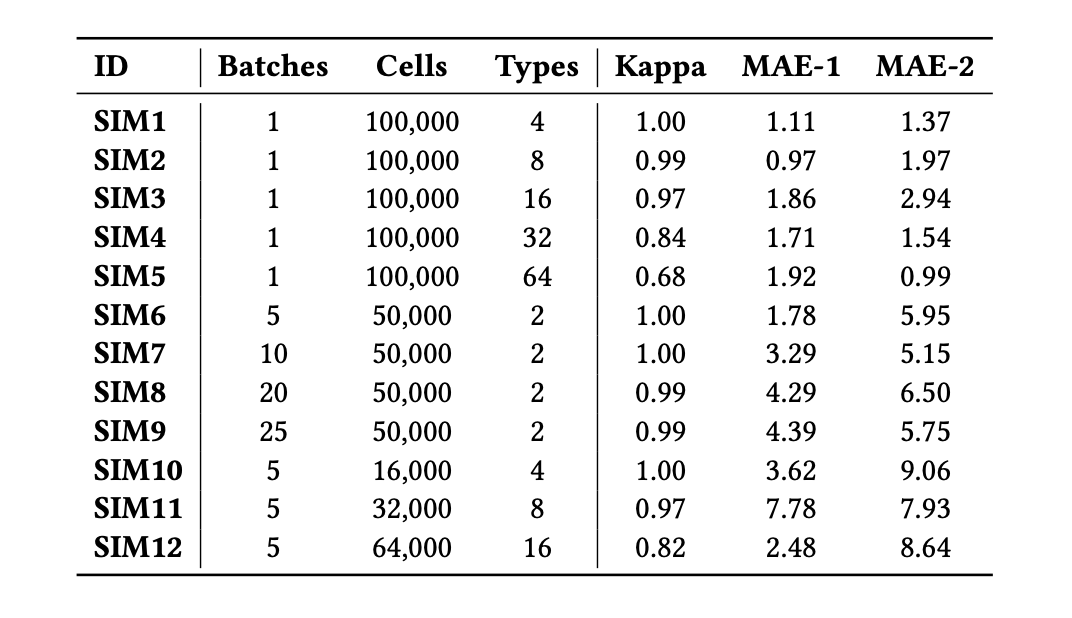
Multi-Target Integration and Annotation of Single-Cell RNA-Sequencing Data
Natalia Khuri, Sapan Bhandari, Esteban Murillo Burford, Nathan P. Whitener, and Konghao Zhao
August 2022, ACM-BCB
Short Paper
▶ Show Abstract
Cells are the building blocks of human tissues and organs, and the distributions of different cell-types change due to environmental or disease conditions and treatments. Single-cell RNA sequencing is used to study heterogeneity of cells in biological samples. To date, computational approaches aided in the discovery of dominant and rare cell-types and facilitated the construction of cell atlases. Integration of new data with the existing reference atlases is an emerging computational problem, and this paper proposes to frame it as a multi-target prediction task, solvable using supervised machine learning. We systematically and rigorously test 63 different predictors on synthetic benchmarks with different properties. The best performing predictor has high Cohen's Kappa scores and low mean absolute errors in single-batch and multi-batch integration experiments.
|
Miscellanea
Awards
-
| Sawyer Price (Wake Forest Computer Science Department) |
May 1, 2024 |
-
| CRA Outstanding Undergraduate Researcher Award Honorable Mention (Computing Research Association) |
December 20, 2023 |
-
| RISS 2023 Summer Scholarship (Carnegie Mellon University Robotics Institute) |
June 1, 2023 |
-
| Wake Forest Research Fellowship (Wake Forest URECA Center) |
May 1, 2022 |
|
|